1.5.1 PROTON AFFINITY
The simplest way to compare the strengths of bases should be to compare the energy released when a base attaches itself to a hydronium ion in gaseous state. The proton affinity (PA) (should be correctly called hydron affinity, but the term ‘proton affinity is used here due to the common usage) of a chemical species X− is defined as the energy released in the reaction
X− (g) + H+(g) → HX(g) PA = − ΔH
where ΔH is the enthalpy change for the reaction. In terms of other calculatable parameters, it can be determined by considering the following sequence of reactions:
Formation of H+ ions: H(g) ⇌ H+(g) + e − ΔHi = I
Formation of X − ions: X(g) + e − ⇌ X − (g) ΔH = – EA
Formation of HX: H+(g) + X−(g) ⇌ HX (g) ΔH = – D
On adding X− (g) + H+(g) → HX(g) PA = − ΔH = + I – EA – D
Here, I = ionization energy of H(g), EA = electron affinity of X(g) and D = dissociation energy of the HX bond. Proton affinities (PA) of ions and molecules of group 14 to 17 elements (in kJ mol−1) are given in Table 1.5.1.
Table 1.5.1 Proton Affinities (PA) of Ions and Molecules
————————————————–
F− Cl− Br− I− H−
1535 1385 1340 1300 1680
OH− SH− SeH−
1567 1450 1420
NH2− PH2− AsH2− CH3−
1645 1640 1560 1690
NH3 H2O HF CH4
375 760 380 675
NH2− O2−
2550 2315
—————————————————-
It is observed that:
- ‘Proton affinities” for all molecules and anions are positive. Hence, a proton or hydron will stick to any species not carrying a positive charge.
- The variations in PA of halide ions are almost entirely due to the variations in the bond dissociation energy of hydrogen halides.
- For iso-electronic systems (NH2−, OH− and F−; or, O2− and Cl−), the proton−proton repulsions due to added nuclear proton charge, outweigh the increased electronegativity of the element, and the PA decreases with the decreasing radius of the anion (decreasing inter-nuclear distance).
- Proton affinities of anions containing the same element with different number of hydrons (protons) is the same as expected due to decreasing proton–proton repulsions:
H2O (760) < OH− (1667) < O2− (2713) (in kJ mol−1)
- The proton affinity of methane (CH4) (675 kJ mol−1) resulting in formation of stereochemically awkward species (CH5+) is higher than that of HF (380 kJ mol−1)
1.5.2 DIFFERENTIATING AND LEVELING SOLVENTS
For determining the strength of an acid HA, a reference base, usually solvent B, is selected and the equilibrium constant K for the following reaction is determined:
HA + B ⇌ BH+ + A−
K = ([BH+][A− ] ) / ( [HA][B] )
or, K [B] = [A–][BH+] / [HA] = Ka
Assuming that the molar concentration of the solvent does not change significantly as a result of its hydronation (protonation) of BH+, Ka can be taken as a constant and is called the ionization constant of the acid HA.1.5
1.5.2.1 Strong and Weak Acids
A strong acid ionizes almost completely in solutions (degree of ionization is almost 1) and has a high Ka, whereas a weak acid has a low degree of ionization and a lower Ka value. If we define
pKa = – log Ka,
then, a stronger acid has a lower pKa and a weaker acid has higher pKa value. Hydrochloric acid HCl, nitric acid HNO3, and sulphuric acid H2SO4 are strong acids, whereas acetic acid, formic acid, oxalic acid, succinic acid, tartaric acid, citric acid, hydrogen fluoride and hydrogen cyanide are weak acids.
1.5.2.2 Leveling Solvent
If a solvent B is such that two acids HA1 and HA2 almost completely (almost 100 %) transfer their H+ ions to the solvent B, then the Ka values for the two acids cannot be distinguished. The solvent B is called the levelling solvent for the two acids. For example, in ammonia as solvent, HCl, HF as well as acetic acid (CH3COOH) are completely ionized to give the NH4+ (BH+) ions:
HA + NH3 → NH4+ + A– (A = Cl, F or CH3COO)
Therefore, ammonia is a leveling solvent for these acids.
1.5.2.3 Differentiating Solvent
In water, hydrochloric acid, hydrofluoric acid and acetic acid ionized to different extents. HCl is almost completely ionized, the pKa ( = log Ka) for HF = 5.2 and for acetic acid = 4.76. Hence, water is a differentiating solvent for these acids.
However, water is a leveling solvent for HCl, HBr and HI, for which acetic acid is a differentiating solvent and gives the acid strength as HCl < HBr < HI:
HX + CH3COOH ⇌ CH3COOH2+ + X– (X = Cl, Br, I)
It can be concluded that acidic solvents (like acetic acid, sulphuric acid or hydrogen fluoride) tends to differentiate between (strong) acids and level off bases, whereas basic solvents like ammonia tend to level off acids and distinguish bases. In fact, all acids and bases stronger than the auto-ionized ions of the solvent, will be leveled off while all the acids and bases weaker than the auto-ionized ions of the solvent will remain in equilibrium and can be distinguished between. Thus in ammonia as solvent, all acids stronger than ammonium ions (NH4+) will be leveled off, whereas in water, all bases stronger than OH– ions will be leveled off.
It can be concluded that acidic solvents (like acetic acid, sulphuric acid or hydrogen fluoride) tends to differentiate between (strong) acids and level off bases, whereas basic solvents like ammonia tend to level off acids and distinguish bases. In fact, all acids and bases stronger than the auto-ionized ions of the solvent, will be leveled off while all the acids and bases weaker than the auto-ionized ions of the solvent will remain in equilibrium and can be distinguished between. Thus in ammonia as solvent, all acids stronger than ammonium ions (NH4+) will be levelled off, whereas in water, all bases stronger than OH– ions will be leveled off.
The strengths of strong acids can be compared by using acidic solvents or aprotic solvents like benzene, which does not assist in ionization of the acids. Both the methods have their own limitations. In aprotic solvents where the solvent does not accept or donate H+ ions, (i) a reference base is required to assist in ionization of the acid, and vice versa; and (ii) the low dielectric constant of the solvent encourages ion-pair formation, so that the results cannot be reliable. Acidic (and basic) solvents, on the other hand, (i) may react with the acids/bases forming complicated products, or (ii) ionization of acid may not be simple, e.g., ionization of nitric acid in sulphuric acid
HNO3 + H2SO4 ⇌ NO2+ + HSO4– + H2O
1.5.2.4 Cosolvating Agents
Cosolvating agents are compounds that react with either H+, A–, B or BH+ and thus increase the ionization of acid HA or base B. Some examples of cosolvating agents are:
- Boron trifluoride as cosolvating agent for hydrogen fluoride in aqueous solutions:
HF + BF3 + H2O ⇌ H3O+ + [BF4] –
- Copper(II) ions (Cu2+) or nickel(II) ions (Ni2+) for ionization of ammonium ions in aqueous solutions;
M2+ + 4NH4+ + 4H2O ⇌ 4H3O+ + [M(NH3)4]2+ (M = Cu or Ni)
- vic-Glycols like ethylene glycol, mannitol, glycerol for boric acid in aqueous solutions:
H3BO3 + (CH2OH)2 ⇌ [(HO)2B{(OCH2)2}]– + 2H2O
[(HO)2B(OCH2)2]– + (CH2OH)2 ⇌ [B(OCH2)4]– + 2H2O
1.5.3 STRENGTH OF HYDRACIDS
The relative strength of the hydracids of group 15, 16 and 17 increases with increasing size of the element (pKa values in parentheses):
H2Te (3) > H2Se (4) > H2S (7) > H2O (14) (Group 16)
HI (–10) > HBr (–9) > HCl (–7) > HF (3) (Group 17)
PH3 (pKa = 27) > NH3 (pKa = 35). (Group 15)
In a group, the acid strength increases with the decreasing electronegativity of the element. This, however, is against the order of the ionic nature of bonds in the hydracids. The ionic character of the bonds is highest for the smallest members in the periodic group (NH3, H2O and HF). This may be due to the following factors:
- The charge density on the conjugate base (anion A–) decreases in the order
F– > Cl– > Br– > I– and HO– > HS– > HSe– > HTe–
Greater charge density will result in greater attraction between the conjugate base and the hydronium ions (H+).
- A larger anion will be more stabilized by solvation effects leading to a higher degree of ionization of the acid HA.
- Smaller anions are bonded to H+ ions through hydrogen bonding also secreasing the ionization of HA.
- Decrease in bond strength of H–A bond with increasing size of A also increases the ionization of HA.
These factors can be better understood by considering the following steps.
The enthalpy of ionization of HX in aqueous solutions, ΔHr, is for the reaction
HX(aq) ⇌ H+(aq) + X–(aq) ΔH = ΔHr
This reaction can be broken down into smaller steps with known parameters:
HX(aq) ⇌ HX(g) ΔH= ΔHv (enthalpy of evaporation of HX solution)
HX(g) ⇌ H(g) + X(g) ΔH= D (H–X bond dissociation energy) \
H(g) ⇌ H+(g) ΔH= I (Ionization energy of H(g)
H+(g) + aq ⇌ H+(aq) ΔH= ΔHaq+ (enthalpy of hydration of H+(g))
X(g) + e –(aq) ⇌ X –(aq) ΔH= – EA (electron affinity of X –)
X –(g) + aq ⇌ X –(aq) ΔH= ΔH aq– (enthalpy of hydration of X–(g) )
This gives ΔHr = ΔHaq++ ΔHaq– + ΔHv + D + I – EA
However, application of this equation for the calculating Ka for the acids presents the following difficulties.
As – log Ka = 2.303 RT ΔG = 2.303 RT(ΔHr – TΔS)
- Determination of solvation enthalpy of the ions (ΔHaq+, ΔHa –) and the evaporation enthalpy of the HX solutions must be approximated by comparing with the molecules which do not ionize.
- Change in entropy (ΔS) can be determined within 2 to 3 units using the equation
ΔS = ΔSHX(aq) – (ΔSH+aq) + ΔSX–(aq) )
The expected values of the different terms involved for some acids are given in Tables 1.6.2 and 1.6.3. The agreement between the calculated and the experimental values is excellent.
Table 1.5.2 Thermodynamic Data for Hydrogen Halides
(kJ mol –1)
——————————————————————————————–
Property HF HCl HBr HI
Dissociation energy (D) 562 430 364 297
Electron affinity (EA) 343 364 343 318
Proton affinity (PA) 1535 1385 1340 1300
Ionization energy of H(g) 1315 1315 1315 1315
Hydration energy of ions (ΔH±) – 1505 –1450 –1425 –1390
Evaporation energy of HX solution – 49 –17.61 –20.9 –23
ΔS (in J deg–1 mol–1) – 88 – 56.5 – 37.6 –13.8
———————————————————————————————-
Table 1.5.3 Thermodynamic Calculation of pK values of Hydrogen Halides at 300 K
————————————————————————————
Acid ΔHr TΔS ΔG pK(calc) pK(obs)
HF – 12.3 – 25 13 2 3
HCl – 57 – 17 – 42 – 7 – 7
HBr – 63 – 13 –50 – 9 – 9
HI – 50 – 4 – 54 – 10 – 10
ΔHr , TΔS and ΔG values are in kJ mol –1
It is observed that:
- ‘Proton affinities” for all molecules and anions are positive. Hence, a proton or hydron will stick to any species not carrying a positive charge.
- The variations in PA of halide ions are almost entirely due to the variations in the bond dissociation energy of hydrogen halides.
- For iso-electronic systems (NH2−, OH− and F−; or, O2− and Cl−), the proton−proton repulsions due to added nuclear proton charge, outweigh the increased electronegativity of the element, and the PA decreases with the decreasing radius of the anion (decreasing inter-nuclear distance).
- Proton affinities of anions containing the same element with different number of hydrons (protons) is the same as expected due to decreasing proton–proton repulsions:
H2O (760) < OH− (1667) < O2− (2713) (in kJ mol−1)
- The proton affinity of methane (CH4) (675 kJ mol−1) resulting in formation of stereochemically awkward species (CH5+) is higher than that of HF (380 kJ mol−1)
1.5.4 STRENGTH OF OXIDO ACIDS
The oxido acids (earlier called oxy acids or oxo acids) are considered as the derivatives of water. A few rules of thumb are given to below to assess the strengths of the oxido acids.
- The strength of the oxido acid increases with the increasing electronegativity of the group replacing the H atom due to the inductive effect of the substituting group: HOH (15.7) < HOI (11) < HOBr ( 8.7) < HOCl (7.4) (pKa values)
- The increase in the oxidation number of atom replacing the H atom of water increases acid strength (oxidation number in parenthesis):
HOCl (I) < HOClO (III) < HOClO2 (V) < HClO3 (VII)
- For uncharged oxido acids in which all the oxygen atoms are attached to a central atom, the acid strength increases with increasing number of =O atoms and decreasing number of hydronated oxygen atoms i.e. OH groups:
O3Cl(OH) > O2S(OH)2 < OS(OH)2 <As(OH)3
- For oxido acids having same composition and oxidation number of the central atom X, the acid strength decreases with increasing size of X atom:
HClO4 > HBrO4 > HIO4
H3PO4 > H3AsO4 > H3SbO4
HNO2 > HPO2 > HAsO2
H2SO4 > H2SeO4 > H2TeO4
Cartledge proposed that the ionic potential ϕ (= 100 × charge / radius (in pm) ratio) may be used to predict the nature of the compound M(OH)n. It will be acidic if ϕ2 > 10, amphoteric if 5 < ϕ2 < 10 and basic if ϕ2 < 5.
Ricci has proposed that
pK = 4 – 9m + 4n.
Pauling proposed that for the acid XOm(OH)n, the acid strength is independent of value of n is given as
m = 0 1 2 3
pK = 7 2 –3 –8
The pK2 is about 5 units higher than pK1 due to electrostatic effects.
- The Formal charge: The formal positive charge developed on X atom due to the formation of coordinate bond (X→O or Xδ+–O δ–) attracts the bonded electron pair between X and OH containing the ionizable H atom. This makes ionization of H+ from the X –O –H bond easier and increases the acid strength of acid. Greater the number of the =O atoms attached to X, greater is the formal charge on X and higher is the acid strength.
- Effect of π bonding: Consider the ionization of a O=X–OH bonded H atom. The negative charge developed on the O atom is stabilized by delocalization of the charge (resonance) involving the π bonded electrons of the X=O bond:
O=X–O–H ⇌ H+ + O=X–O– ↔ –O–X=O ≡ δ–O–X–Oδ–
Larger the number of X=O bonds, more is stabilization as delocalization involves more atoms. As a result, the charge on each O atom in XOn– as n increases. It is obvious that the charge on the O atoms in XO–, XOn–, XOn– and XOn– is –1. –(1/2), –(1/3) and –(1/4) respectively:
HXO ⇌ H+ + XO–
HXO2 ⇌ H+ + XO2–(1/2)
HXO3 ⇌ H+ + XO3–(1/3)
HXO4 ⇌ H+ + XO4–(1/4)
- Ion-pair Formation: Due to the electrostatic attraction between the anions and the cations in solutions, ion pairs are formed, which reduce the effective ionization of the acids and lower the acid strength. Due to reduced charge on the XOn–. the ion pair formation with H+ ions decreases in increasing number of n, and the acid strength increases regularly from XO– to XO4–.
- Significance of d Orbitals: The differences between the acid strengths of oxido acids of second period and corresponding acids of the third and hihger periods (HNO3 vs HPO3 or HAsO3) is due to (i) relative increase in size of central atom, (ii) lower stability of (p–p-π) bonds due to more diffused character of p ornitals, and (iii) tendenency of heavier atoms to accept electrons in their low energy d orbitals forming M –O (p→d-π) bonds. Bond strength data indicates that the (p–p-π) bond character increases from 54% for ClO3– to 37% for BrO3– and to 21% for IO3–.
- Solvation Effects: In addition to formal charge and delocalization effects, solvation of X=O by water is acid strengthening, whereas the solvation (hydration) of X–OH is acid weakening due to the inductive effect of water molecules:
H–O–X=O∙∙∙∙Hδ+–Oδ––H O=X–O–H∙∙∙∙ δ–OH δ+2
The positively charged H atom of water molecule attracts the electrons of the acid towards itself (I) and weakens the O–H bond increasing its ionization, whereas the hydration of OH group is by forming H∙∙∙∙O bond in which the ionizable H atom gets trapped and its ionization decreases. This weakens the acid strength.
The pK1 values of H3PO4 (2.1), (CH3O)PO3H2 (1.52) and (CH3O)2PO2H (0.76) show that the solvation of ionizable OH group has an acid weakening effect.
1.5.5 STRENGTH OF ORGANIC ACIDS
The organic acids have either a carboxylic group of OH group capable of ionizing in solutions and releases hydronium ions (H+). The effect of substituent in organic acids can be of two types: (a) inductive and (b) delocalization or resonance.
1.5.5.1 INDUCTIVE EFFECTS
Inductive effects are important for aliphatic unconjugated systems where delocalization of electrons is not possible.
Substitution by groups which are more electronegative than carbon increases strength of aliphatic acids, whereas substitution by groups which are less electronegative than carbon decreases their strength as is seen by the pK values the acids:
H––CH2COOH 4.76 MeO–CH2COOH 3.53
F–CH2COOH 2.66 Me3Si–CH2COOH 5.22
Cl –CH2COOH 2.86 MeS–CH2COOH 3.72
I–CH2COOH 2.66 Me3C–CH2COOH 5.05
This can be understood by the electrostatic model of the molecule. If X is more electronegative than C, the bonded electron pair between the X–C bond is polarized to give a slightly positive charge on carbon atom. This then attracts the electron pair from the adjacent bond (C–OH), weakens the bond and and results in easier ionization of H+ ions. This is called the inductive effect. The polarization of bonds due to inductive effect is a weak effect and does not extend to more than 2 or 3 atoms. Thus the pK of Me3C–CH2CH2COOH is the same as that of acetic acid.
1.5.5.2 Field Effects
The field effect is the interaction of dipolar C–X bond directly with the ionizable hydronium ion through dielectric of molecule itself. Though the inductive effect is internal bond polarization only, as an approximation, is includes both the effect.
1.5.5.3 Electron Withdrawing and Electron Releasing Groups
Groups that increase the positive charge on adjacent atom are called electron withdrawing groups and are said to have a +I effect. Groups that decrease the positive charge or increase the negative charge on adjacent atom are called electron releasing groups and are said to have a –I effect.
The increasing electron withdrawing power is observed to increase with:
- Oxidation number of central atom:
MeS– < MeSO– < MeSO2–
CH3 < CH2=CH– < CH≡C–
Me3N– < –N3 < –NO < –NO2
–I < –IO < –IO2
- Increasing s character of the hybrid orbitals used in bonding:
CH3 < CH2=CH– < CH≡C
–CONH2 < –CN
From the ionization constants of substituted acetic acids, the order of different substituents with increasing electron withdrawing power is:
Me3C– < Me2CH– < MeCH2 – < H < CH2=CH– < C6H5 < CH3S–
< CH2Cl– < C2H5C(O)O– < I– < N3– < –CF3 < Br– < Cl–
< C6H5SO– < –CN < CH23SO2– < –NO2
1.5.6 EFFECT OF DELOCALIZATION OF ELECTRONS (RESONANCE or MESOMERIC EFFECT)
Delocalization or resonance is more important for aromatic compounds and for aliphatic compounds with alternate double and single bonds, where inductive effects have almost no significance, We consider a few examples.
1.5.6.1 Nitrophenols
Nitro group is an electron withdrawing group. Its inductive effect will increase the acid strength of nitrophenols in the order ortho > meta > para. But due to delocalization of electrons, weakening of O–H bond will be maximum in the para position (Figure 1), less for ortho position and nil for the meta position.
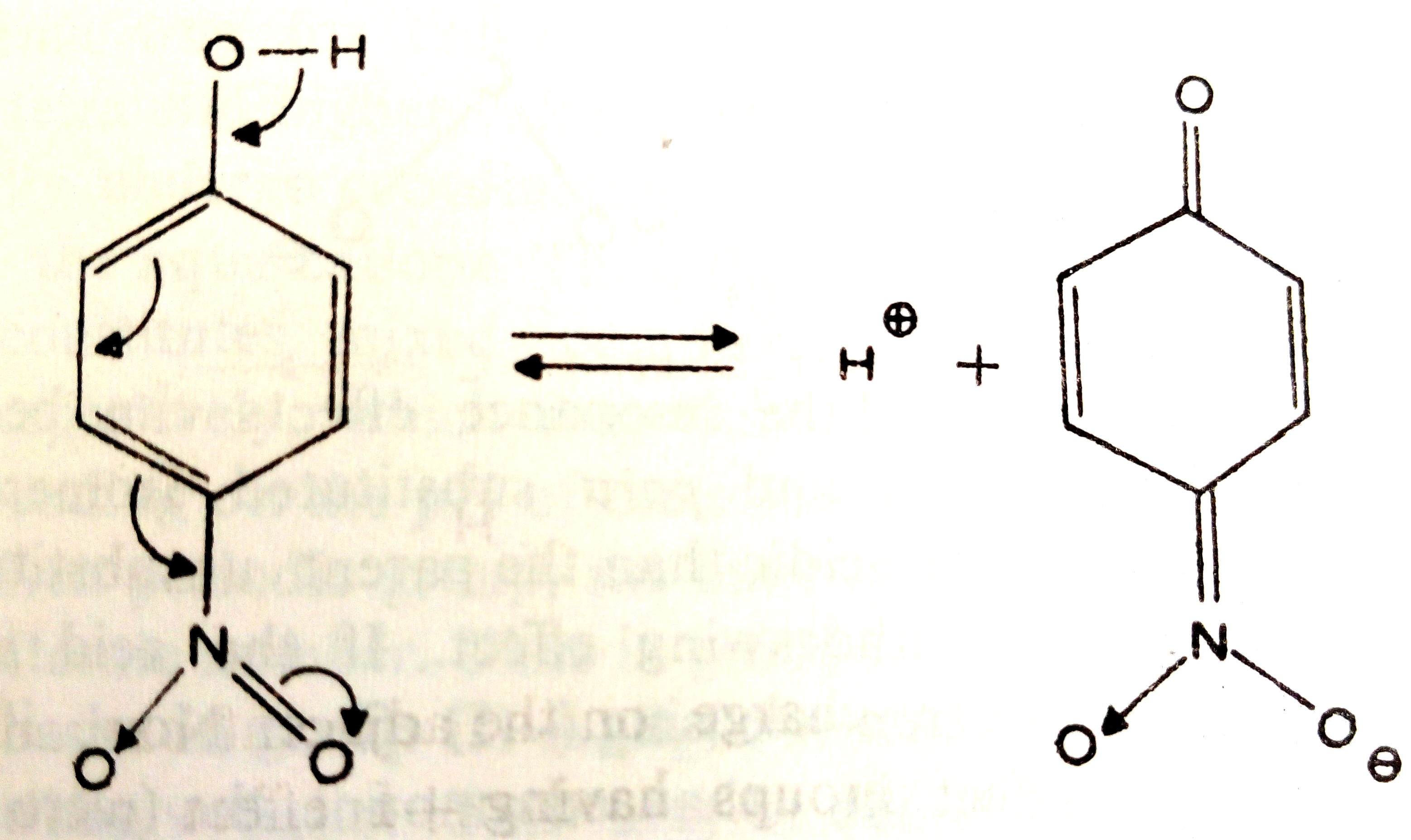
Figure 1 Ionization of p-nitrophenol
Hence, the acid strength of nitrophenols is in the order:
unsubstituted < meta < ortho < para.
1.5.6.2 Trimethylsilylanilines
Due to the delocalization of the electron pair on the NH2 group, the SiMe3 group exterts a strong electron withdrawing effect on the NH2 group from ortho and para positions. The silicon aton accepts the electron pairs in its 3d orbitals (Figure 2). It introduces a weak acid weakening effect from the meta position (Si is less electronegative than C atom). Hence, the acid strength increases (and the base strength decreases) in the order
para > ortho > unsubstituted < meta.
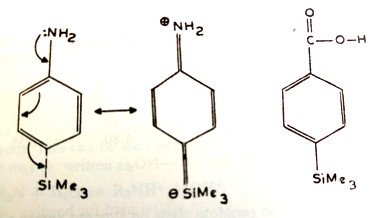
Figure 2 Ionization of p-trimethylsilylaniline and absence of
delocalizationof the OH bond with the aromatic ring
There is no delocalization of electron pair of O–H bond and the aromatic ring, so that the mesomeric effect of the trimethylsilyl group in any position. Therefore it shows only a weak electron withdrawing inductive group ( Si is less electronegative than C atom).
1.5.6.3 Methoxybenzoic Acids
In the meta position, the methoxy (OMe) group (correctly called methoxide, IUPAC, 2005, Red Book) has a slight acid strengthening inductive effect. However, methoxy group is a strong electron releasing effect due to delocalization (resonance) and leads to decreased acid strength from the para and ortho positions. (In the meta isomer, the COOH group is not involved in delocalization.)
In actual compounds, three types of trends are observed:
- If the meta substituted benzoic acid is more acidic than the benzoic acid, then the group has electron withdrawing effect. It is said to have a –I If the para isomer is more acidic that the meta isomer (as in nitro phenols), the inductive and delocalization effects operate in the same way.
- If reversal takes place and meta substituted compounds are less acidic than the para compounds (vice versa for base strength), then the inductive and delocalization effects operate in the opposite directions (methoxybenzoic acids and trimethylsilylamines.
- If the meta substituted compounds are more acidic than the pare substituted compounds, there is no effect of delocalization of electrons involving the substituent.
Substituted amines of phenols are best for studying the effect of delocalization effects due to electron withdrawing group, whereas benzoic acids are best for electron releasing groups.
Some substituents like unsaturated groups or phenyl rings, can become electron withdrawing groups in one situation and electron releasing in other due to delocalization or mesomeric effects. This is shown by pK values of substituted phenols and puridines:
Substituent H m-C6H5 p-C6H5
Phenols 9.95 9.59 9.51
Pyridinium ion 5.11 4.80 5.55
1.5.7 OXIDES
An oxide is called acidic or basic depending upon its behavior in solutions. Acidic oxides increase the concentration of H+ ions whereas basic oxides decrease its concentration.
MO + HOH ⇌ H+ + MO2– (Acidic Oxide)
MO + 2H+ ⇌ M2+ + H2O (Basic Oxide)
Amphoteric oxides act as acidic or basic depending upon the pH of the solutions:
Al2O3 + 6H+ ⇌ 2Al3+ + 3H2O (Basic nature)
Al2O3 + 2OH– ⇌ 2AlO2– + H2O (Acidic nature)
Some amphoteric oxides are those of Be(II), Al(III), Ga(III), Sn(II), Sn(IV), Pb(II), Pb(IV), As(III), Sb(III), Ti(IV), V(III), V(V), Cr(II), Cr(III), Fe(II), Fe(III), Co(II), Ni(II), Cu(I), Cu(II), Zn(II), Cd(II), etc.
- The bivalent metal oxides are more basic than the higher valent oxides. The high valent oxides are not really amphoteric as they undergo coordination reactions with anions rather than form hydrated aqua cations. The ions written usually as VO2+ and VO2+ are actually mixed anion complexes like [V(OH)2(OH)2]2+ and [V(OH)4(H2O)]+ respectively, which may further condense to form polynunlear complexes depending on the pH of the solution.
- In a periodic group, the basic nature of the oxides increases with increasing atomic number. For example, in group 12, BeO is amphoteric, heavier oxides (MgO, CaO, SrO and BaO) are basic. In group 13, B2O3 is acidic, Al2O3 and Ga2O3 are amphoteric, while In2O3 is basic. This may be due to higher charge density on the smaller cations in the oxides, which polarizes the oxide ions (Fajan’s rule: smaller cations have a higher charge density which polarizes the anions and tends to form covalent bonds with it.)
- It is possible that all oxides tend to be amphoteric, but the pH ranges may be insufficient to show the complete behavior. For example, N2O5 in aqueous solutions shows acidic behavior at all pH ranges and forms NO3– However, in sulphuric acid medium, it ionizes to give NO2+ (nitronium) ions:
N2O5 + H2O → 2H+ + NO3– (in aqueous solutions)
N2O5 + H2SO4 ⇌ 2NO2+ + H2O + HSO4– (in sulphuric acid medium)
- The metal oxides are more basic than the corresponding hydroxides which they form on hydrolysis in aqueous solutions:
O2– + H2O → 2OH–
The metal oxides may form many salts in molten states which rapidly decompose in aqueous solutions:
Li4PbO6 + 3H2O → 4LiOH + Pb(OH)2
Ba3MoO6 + 3H2O → 3Ba(OH)2 + MoO3.
1.5.8 BASIC STRENGTHS OF AMINES
Solvation Effects: As the acid and basic strengths are usually determined in aqueous solutions, it becomes important to consider the effect of solvation (aquation in aqueous solutions). Consider the basic nature of alkyl amines:
R3N + H2O ⇌ R3NH+ + OH– .
Addition of a bare hydronium ion (H+) presents no steric difficulty because the ions has a very very small size. Thus, the base strength of alkyl amines in gas phase follow the expected order from the electron releasing inductive effecr of alkyl groups:
NH3 < RNH2 < R2NH < R3N
The solvation of amines through hydrogen bonding tends to increase the base strength proportional to the extent of solvation (Figure 3).

Figure 3 Hydration of (a) t-trilkylamine and (b) hydration of alkyl amine.
Larger the number of H atoms on amine, more is hydration through hydrogen bonding with water
The solvation energy decreases with the number of alkyl groups present and is in the order:
NH3 > RNH2 > R2NH > R3N
Because of the opposing trends, the maximum basic strength is observed at secondary amines (R2NH).
Effects of Substituents: Replacement of H atoms by electron releasing groups (alkyl, methoxide OMe, or hydroxide) increases its base strength as shown by their pKb values.
Kb for ammonia (NH3) = 4.74
Electron withdrawing groups
NH2NH2 5.77 NH2OH 7.87
Electron releasing groups
MeNH2 3.36 Me2NH 3.29 Me3N 4.28
EtNH2 3.25 Et2NH 2,90 Et3N 3.25
i-PrNH2 3.28 i-Pr2NH 1.95
t-BuNH2 3.52 t-Bu2NH 3.32 t-Bu3N 3.58
On the other hand, replacement of H atoms by electron withdrawing groups (halogens, nitro, cyano, etc.) decreases its base strength of ammonia. In water, ammonia is a base, but nitrogen trifluoride (NF3) does not show any basic behavior. In fact, NF3 gives acidic aqueous solutions due to hydrolysis (hydrolysis is complicated and reaction not gives here). This may be due to high electronegativity of fluorine which withdraws the electron density from nitrogen atom making it slightly positive.
Steric Effects: In addition to the solvation effects from dialkyl to trialkyl amines, steric effects may also become important. For accomodating bulkier groups in, say, t-trimethylbutylamine (t-Bu3N), the nitrogen atom may be forced to use the sp2 hybrid orbitals, leaving the electron pair in the symmetrical sp hybrid orbital, which is not suited for bonding with hydronium ion (Figure 4). This is called the back strain or B-strain. (The electron pair on nitrogen atom in ammonia is present in the strongly directional sp3 hybrid orbital.)

Figure 4 As size of alkyl group increases, a back-strain (B-strain) is developed and the lone pair on N atom of amine is forced to occupy s orbital as the alkyl group becomes very large or bulky
Steric effects may not be very important for smaller groups like methyl or ethyl, for which solvation effects are more important.